【コラム】
免疫染色用の細胞サンプルの準備(固定編)
免疫染色用の細胞サンプルの準備(固定編)
細胞サンプルの準備は組織、細胞染色において、目的のタンパク質の局在や染色性を左右する非常に重要なステップとなります。固定、透過処理、ブロッキングを間違うと、染色に失敗したり誤った染色になったりする恐れがあります。
ここでは細胞サンプルを準備するにあたり、固定のステップで注意すべきことをまとめています。
細胞サンプルを用意する前の注意
まず参考文献などから目的のタンパク質が細胞のどこに(局在性)どうやって発現しているのか確認する必要があります。もし目的のタンパク質が通常の状態では発現されていないのであれば、前処理として、その細胞を他のタンパク質や化学物質で刺激して、目的タンパク質の発現を誘導させます。
細胞染色では、細胞密度(コンフルエント)も大切であり、通常、60 ~ 80% 程度が細胞染色に適しています。細胞間の連絡を保つためにもこの程度の細胞密度が重要となります。細胞間結合マーカーなどの観察には細胞密度が不適切なことにより致命的となる場合があります。
また、細胞数が少なすぎると、広い視野で探すために低倍率での観察となり、十分な細胞数を確保するのに何枚も撮影しなければならなくなります。一方で、細胞が多い、つまり密度が高ければよいというわけでもありません。多すぎる細胞は、細胞構造がわかりにくくなります。例えば、満席のスタジアムは、空席が多い状況よりも、ひとりひとりが分かりにくくなるイメージです。
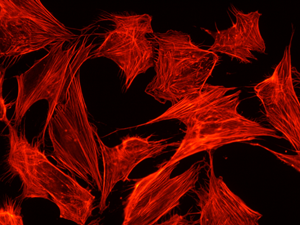
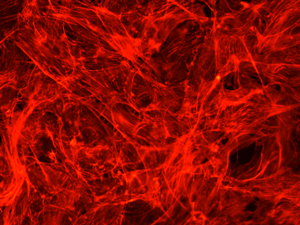
MC3T3-E1 細胞の細胞骨格を Rhodamine 標識 Phalloidin で染色
細胞数が適切(細胞密度 70%)
細胞数が多すぎ(細胞密度 100%)
細胞はカバーガラス上?スライドガラス上?どちらで観察がよいか?
普段、細胞はシャーレや培養フラスコで培養しているかと思いますが、染色に用いるときには、一定の厚みのあるガラス上に乗せなければいけません。
ガラスはカバーガラスかスライドガラスを用います。これは、観察する細胞の種類が接着細胞、または非接着細胞であるかによって選ぶのですが、それぞれどちらのガラスを使うかわかりますか?これからそれぞれの細胞を準備する方法をご説明します。
接着細胞
接着細胞は、ガラスやプラスチックの表面に張り付いて増殖します。そのため、培養シャーレなどにガラスをいれて、その表面上に張り付かせるのですが、スライドガラスだと大きすぎて小さなウェルには入りきらず、また厚みがありすぎて、ガラス表面にうまく均一に広がって増えてくれないかもしれません。また、スライドガラス一面に細胞が広がっては、染色するのも大変です。そのため、接着細胞にはカバーガラスを無菌的に培養プレートやウェルプレートに入れて、その上で細胞を培養して観察します。
カバーガラスはオートクレーブやクリーンベンチ内の UV 照射で滅菌しておきます。使用前に、クリーンベンチ内でエタノール洗浄し、乾燥させてから培養プレートやウェルプレートに入れます。もし細胞がカバーガラスに接着しにくい場合は、細胞がくっつく足場となる、細胞外基質コーティング剤を使うことをお薦めします。細胞の種類によって最適な細胞外基質コーティング剤は変わりますが、ポリ-L-リシンはほとんどの細胞に使うことができます。
| 細胞の種類 | 推奨細胞外基質コーティング剤 |
|---|---|
| 胚性幹細胞(ES 細胞) | ラミニン |
| 初代ケラチン生成細胞 初代肝細胞 |
コラーゲン |
| 微小上皮血管細胞 | ゼラチン |
| 神経幹細胞 | フィブロネクチン |
非接着細胞
ガラス上で培養しなくともよい非接着細胞は、他の容器で培養した細胞をスライドガラス上に滴下して観察できます。染色するためには、スライドガラスに乗せる前に、まず、固定剤を培養液に直接入れて固定します。理由としては、スライドガラス上に移して、細胞の状態が変わってしまうと、本来見たいタンパク質が無くなったり、細胞の形が変わってしまったりするので、スライドガラスに移す前に、細胞の形、状態を"固定"してしまいます。
大まかな目安ですが、1 × 106 の細胞に対して、1 mL の 4% パラホルムアルデヒド(PFA)を加えます。20 分静置したら、遠心洗浄で固定剤を取り除いて、2 回蒸留水で遠心洗浄します。そのあと、蒸留水で再浮遊させた細胞をゼラチンコートされたスライドガラスにピペットチップなどを使ってスメアをつくります。

固定テクニックの重要性
固定は細胞形態、細胞の全体性、細胞構造を維持するのに大切なステップです。固定することで、タンパク質分解酵素による細胞の自己融解や細胞サンプルの腐敗を防ぐことができ、さまざまな染色過程で細胞自体が壊れないようにします。ですので、できる限り自己融解や腐敗を防ぐために、細胞は培養液から出したらすぐに固定することが大切です。また、固定が不十分の場合、目的の抗原が壊れたり、本来その抗原がない場所に流れてしまったりします。以下に一例を示します。
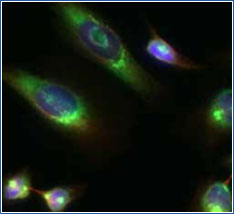
悪い例
良い例
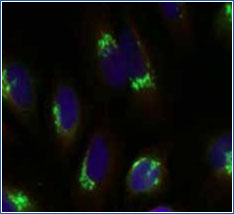
この二つの画像はそれぞれ、HeLa 細胞を、GM130/GOLGA2 というゴルジ体のマーカー(目印)で染色したものであり、同じ細胞です。
左図は失敗した例です。GOLGA2 の染色されている部分が細胞質にも見られます。本来は右図のようにゴルジ体の膜部分にのみ限局して見られるものになります。
固定がきちんとできていないと結果が正しいのかを判断することができません。
主な固定剤
アルデヒド系固定剤
ホルマリンやホルムアルデヒドのようなアルデヒド系は、細胞構造を維持させるなど、膜タンパク質の免疫染色に向いています。この固定剤は、タンパク質同士をフリーのアミン類を介して架橋し、分子間架橋によってタンパク質同士をネットワークのようにつなげてしまいます。ただ、この架橋結合は、固定時間が長くなると、抗原を隠したり、抗原性を弱めてしまったりします。細胞であれば、4% PFA か 10% ホルマリン溶液に室温で 10 ~ 20 分程度が適しています。
有機溶媒/酸系固定剤
メタノール、アセトン、ピクリン酸のような有機溶媒は強力な脱水作用により、細胞内のタンパク質の沈降を起こします。細胞構造の維持もできますが、脱水作用により組織を収縮させたり、小さな溶解性の分子や脂質は取り除かれてしまいますので、一般的に電子顕微鏡用細胞サンプルの固定には不向きです。さらに、沈降固定剤は過剰発現させた蛍光タンパク質(GFP など)を変性させてしまいますので、そのようなタンパク質を持つ細胞サンプルの固定にも向きません。一般的には冷蔵保存、できれば -20℃ で冷やしておいたメタノールを用いて、4℃ で 10 ~ 20 分程度、細胞を固定します。アセトン固定はメタノールよりやや穏やかな作用であるため、冷蔵保存してあるメタノールとアセトンを 1:1 で混ぜた固定液を使用することもあります。メタノールやアセトンで処理した細胞サンプルは、透過処理(抗体が細胞膜を通過できるようにする処理)はしなくても問題ありません。
10% ホルマリン溶液と 4% PFA の違い
PFA は重合したホルムアルデヒドの粉末で、使用前に PBS で溶解します。ホルマリンは飽和ホルムアルデヒド溶液(37% w/v)で、ホルムアルデヒドが重合しないようにメタノールが安定剤として加えてあります。3.7% PFA 溶液が 10% ホルマリン溶液に相当します。
固定剤の利点・欠点の比較
ホルムアルデヒド
利点
汎用的な固定剤、初めて使う抗体や抗原がある場合は、まずホルムアルデヒドを試すことをお薦めします。細胞構造の維持や、細胞膜上タンパク質の固定に優れています。
欠点
抗体によっては、ホルムアルデヒド固定の抗原に結合できなくなります。抗原の過剰な架橋結合によって、シグナルが弱くなることもあります。
メタノール
利点
アルデヒド系が使えないエピトープ、抗原にも使えます。透過処理が不要なのも利点です。
欠点 抗体によっては、メタノール固定の抗原に結合できなくなります。高揮発性で可燃性なので、保管に要注意です。過剰発現した蛍光プローブには適していません。
アセトン
利点
アルデヒド系もメタノールも使えないエピトープ、抗原にはアセトンが使えます。これも透過処理は不要です。メタノールより少し穏やかな固定方法になります。
欠点
抗体によっては、アセトン固定の抗原に結合できなくなります。高揮発性で可燃性なので、保管に要注意です。過剰発現した蛍光プローブには適していません。
以上、免疫染色用の細胞サンプルを準備するにあたり、固定ステップで気を付けるポイントやコツを解説しました。染色はちょっとしたことで仕上がりに大きな差がでます。器用さも問われるところですが、やはり基本的な手技順序を守ることが第一です。
関連コラム

※掲載内容 2021 年 6 月現在の情報に基づいています。